MIT Launches Open-Source AI Model Boltz-1 for Biomolecular Predictions
Scientists from the Massachusetts Institute of Technology (MIT) have unveiled Boltz-1, a new open-source AI model that promises to advance biomolecular structure prediction and drive progress in biomedical research and drug development. The model is designed to provide a powerful tool for researchers working to understand the complex structures of proteins and other biomolecules.
A Breakthrough in Biomolecular Modeling
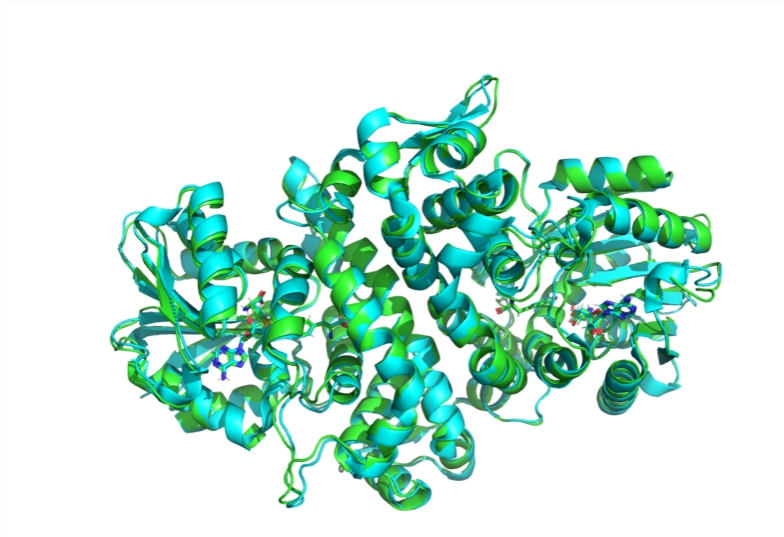
Boltz-1 is the first fully open-source model to achieve the same level of performance in predicting biomolecular structures as Google's AlphaFold3, which is considered a significant advancement in the field. The model is developed by the MIT Jameel Clinic for Machine Learning in Health, with contributions from graduate students Jeremy Walwender and Gabriel Colso, along with researchers Saro Pasaro, and professors Regina Barzilay and Tommy Akala.
At the launch event on December 5, Walwender and Colso explained that Boltz-1 is intended to be a collaborative platform to foster global cooperation in scientific discovery. Colso emphasized the importance of community involvement, stating that the model’s name, oltz-1 was specifically chosen to encourage participation from researchers worldwide.
The Importance of Protein Structure Prediction
Proteins play a crucial role in virtually every biological process, and their function is intrinsically linked to their three-dimensional structure. Understanding how proteins fold and their resulting structures is key to designing new drugs and engineering proteins with specific functions. However, accurately predicting these structures has been a longstanding challenge due to the complexity of the folding process, where long chains of amino acids fold into intricate 3D shapes.
AI models like AlphaFold2 have revolutionized the field by using machine learning to predict protein structures with remarkable accuracy. AlphaFold3 further refines this by employing generative AI models, but it has faced criticism for not being fully open-source, limiting its accessibility to the broader research community.
Boltz-1: A More Accessible Model
In response to these concerns, MIT's research team developed Boltz-1, building on the ideas behind AlphaFold3 while making the model open-source. The goal is to provide the scientific community with a robust, efficient tool for biomolecular modeling that can be freely accessed and utilized. After conducting multiple experiments over four months, the MIT team demonstrated that Boltz-1 performs just as well as AlphaFold3 in complex biomolecular predictions.
The team also overcame challenges related to the ambiguity and heterogeneity present in the Protein Data Bank, an important repository of biomolecular data. Their solution improves the model’s accuracy and provides researchers with a reliable method for structure prediction.
Invitation for Collaboration
Looking ahead, the MIT researchers plan to continue enhancing Boltz-1's performance, particularly with the aim of reducing prediction times. They are actively inviting researchers to test the model and contribute to its ongoing development through GitHub and a Slack channel. By fostering a collaborative environment, the team hopes to inspire creative applications and further innovation in the field of biomolecular research.
For more information, visit MIT's Jameel Clinic Boltz-1 Project.
Key Points
- Boltz-1 is the first fully open-source model for biomolecular structure prediction, offering performance comparable to AlphaFold3.
- The model is designed to promote global collaboration and accelerate progress in biomedical research and drug development.
- MIT researchers aim to make protein structure prediction more accessible to the broader scientific community by providing an open-source alternative.
- The MIT team is inviting researchers to participate in improving and utilizing the model through GitHub and Slack.